


Shandong Provincial Hospital for Skin Diseases & Shandong Provincial Institute of Dermatology and Venereology, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan, Shandong, China
Epidermolysis bullosa encompasses a group of inherited blistering skin disorders. The pathogenic mutations in 10–25% of patients with epidermolysis bullosa have not been identified by Sanger sequencing. The aims of this study were to identify the pathogenic sequence alterations in a large cohort of Chinese patients with epidermolysis bullosa and to clarify the relationship between clinical phenotypes and genotypes. Whole-exome sequencing was performed on 44 pedigrees and 13 sporadic cases. The results were further confirmed by Sanger sequencing. In total, 52 mutations, comprising 19 novel and 33 previously reported mutations, were identified in 5 genes, with a mutation detection rate of 100%. A relationship between subtypes and pathogenic genes was established: 12 cases of epidermolysis bullosa simplex were associated with mutations in KRT5/14 and PLEC; one case of junctional epidermolysis bullosa carried mutations in ITGB4; and 44 cases of dystrophic epidermolysis bullosa were caused by mutations in COL7A1. The results of this study support whole-exome sequencing as a promising tool in the genetic diagnosis of epidermolysis bullosa.
Key words: epidermolysis bullosa; mutation; whole-genome exon sequencing; KRT5; KRT14; COL7A1.
Accepted May 26, 2021 Epub ahead of print May 27, 2021
Acta Derm Venereol 2021; 101: adv00503.
doi: 10.2340/00015555-3843
Corr: Hong Liu, Shandong Provincial Hospital for Skin Diseases & Shandong Provincial Institute of Dermatology and Venereology, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan, Shandong, China. E-mail: hongyue2519@hotmail.com
Epidermolysis bullosa is a rare inherited skin disorder. According to previous reports, immunofluorescence mapping established the epidermolysis bullosa subtype in 76% of cases, while the molecular pathology was identified in 90% of cases by the targeted next-generation sequencing panel. The detection rate of pathogenic genes and mutations in 57 Chinese patients with epidermolysis bullosa in this study was 100% by whole-exome sequencing. A total of 52 pathogenic mutations were found in 5 genes, of which 25 were new mutations. The phenotype-genotype correlation was established in all cases. The results of this study suggest that whole-exome sequencing improves genetic diagnostic sensitivity in epidermolysis bullosa.
Epidermolysis bullosa (EB) is a hereditary skin and mucosal membrane disease characterized by blisters in response to mild mechanical trauma. The inheritance pattern of EB is autosomal dominant (AD) or autosomal recessive (AR). Clinical manifestations of EB vary from mild symptoms, including blistering, scarring, and erosion on the hands and/or feet, to severe symptoms presenting extensive mucocutaneous blisters at birth (1). The worldwide incidence and point-prevalence of EB are 1.4–41.3 per million live births and 2.8–54.0 per million population, respectively (2). EB is classified into 4 major types: EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB), and Kindler EB (3). Diagnosis of EB can be confirmed by: (i) immunofluorescence mapping (IFM), based on the level of skin cleavage; (ii) transmission electron microscopy (EM), through the analysis of skin ultrastructure; (iii) direct genetic analysis of genes associated with EB by Sanger sequencing (SS); and (iv) in cases without a clear candidate gene, or where candidate genes have been ruled out, or in cases when SS was the first chosen method and did not identify the pathogenic variant, targeted next-generation sequencing (NGS) or whole-exome sequencing (WES) is recommended (1, 4).
To date, 16 genes with mutations have been reported in the pathogenesis of EB, indicating the genetic heterogeneity of EB. EBS is the primary type of EB, which frequently presents mutations in KRT5, KRT14, and PLEC; JEB is usually associated with mutations in LAMA3, LAMB3, and LAMC2; and DEB and Kindler EB are caused by mutations in COL7A1 and FERMT1, respectively (3). Currently, the causal genes of approximately 10–25% of EB patients remain unidentified by SS (5–8).
NGS has shown promising improvement in the diagnosis of EB. In a laboratory study in Germany, the discovery percentage of EB on a skin biopsy by IFM and a targeted NGS multi-gene panel was 76% and 90%, respectively (9). Compared with skin microscopy and SS, WES is a more sensitive technique, which can refine and improve the diagnosis of EB, particularly in mild cases (10). In 2014, Takeichi et al. (10) performed WES and identified genetic mutations in 9 cases of EB in which a genetic diagnosis failed using common diagnostic methods.
The aims of this study were to further explore the value of WES in genetic diagnosis of EB and the relationship between EB phenotypes and genotypes in the Chinese Han population. Comprehensive sequencing analysis was performed using WES in a cohort of 44 pedigrees and 13 sporadic patients from the Chinese Han population.
Subjects
In total, 44 pedigrees, including 71 patients with EB and 60 healthy family members, and 13 sporadic cases with a clinical and histological diagnosis of EB were recruited to this study. The diagnosis of EB was based on patients’ clinical information and skin biopsies. A total of 364 in-house controls were recruited as controls for screening mutations.
This study was approved by the institutional review board of the Shandong Provincial Institute of Dermatology and Venereology.
Whole-exome sequencing
With written consent, patients’ DNA was extracted from 5 ml peripheral blood using the TIANamp Blood DNA Midi Kit (Tiangen Biotech, Beijing, China). The quantity of DNA samples and the purity of nucleic acids were detected on a Nanodrop 8000 (Thermo Scientific, Waltham, MA, USA). A total of 57 probands and 364 controls were sequenced by WES. Whole-exome capture was performed by in-solution hybridization using a Biorupter to acquire 150–200-bp fragments. High-throughput sequencing was performed by massively parallel sequencing reads on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA). The analysing software FastQC (Babraham Bioinformatics, Cambridge, UK) was used to remove low-quality reads. The resulting reads were mapped to the human genome reference hg19 by Burrows-Wheeler Aligner. The Genome Analysis Toolkit (GATK Best Practices (Broad Institute, Cambridge, MA, USA)) was used for debugging base quality scores, realigning indels, and removing duplicates. The GATK VariantRecalibrator and ApplyRecalibration commands with the parameter ‘’--ts_filter_level 99.0.’’ were used to recalibrate variant scores.
Sanger sequencing
Mutations identified by WES were verified in all patients with EB and healthy family members by SS. Primers were designed on the NCBI website (National Center for Biotechnology Information, www.ncbi.nlm.nih.gov), PCR was used for amplification, and sequencing was performed as described previously (11). All amplicons were sequenced directly on an automated DNA sequencer (3500xL Dx Genetic Analyzer; Applied Biosystems, Waltham, MA, USA).
Mutation analysis
Non-synonymous single nucleotide variants (SNVs), frameshift mutations, or splice variants located at the splice region of EB pathogenic genes were considered. Pathogenic mutations were considered as: (i) protein truncating variants; (ii) registered as pathogenic in the Human Gene Mutation Database (HGMD), ClinVar, LOVD3, or disease-specific databases, or met the criteria of American College of Medical Genetic (ACMG); or (iii) having an allele frequency of 0% in databases of the East Asian population, such as 1,000 Genomes, ExAC, and gnomAD, a SIFT score of less than 0.025, and a Polyphen-2 and CADD score greater than 0.95 and 10, respectively.
Subjects
A total of 44 pedigrees, 13 sporadic cases, and 364 healthy controls were included in the study. Twenty-two EB probands were female and 35 were male, age range 1–58 years (mean age 20.05 years), and the onset age was from birth to 31 years (mean age: 4.37 years). Of the 364 healthy controls, 164 were males and 200 were females at a mean age of 44.8 years. All participants were Han Chinese.
In total, 52 mutations, comprising 19 novel and 33 previously reported mutations, were identified in 5 genes, with a mutation detection percentage of 100% (Table I). Five genes were found; KRT5 (NM_000424), KRT14 (NM_000526), PLEC (NM_201384), ITGB4 (NM_000213) and COL7A1 (NM_000094). These mutations were further confirmed by SS (Fig. S1), and were not found in any controls. All mutations were considered as pathogenic or damaging, based on the criteria of pathogenic mutations described in the Methods.
Table I. Clinical phenotypes and mutations identified in patients with epidermolysis bullosa (EB) in this study
Based on the clinical manifestations and genetic sequencing results, 57 probands were divided into 3 subtypes; 12 EBS, 1 JEB, and 44 DEB. The clinical and genetic characteristics are summarized in Table I. The landscape of all genetic alterations of these 5 genes in the Chinese population is presented in Fig. 1 by searching PubMed and the CNKI Library (until May 2020).
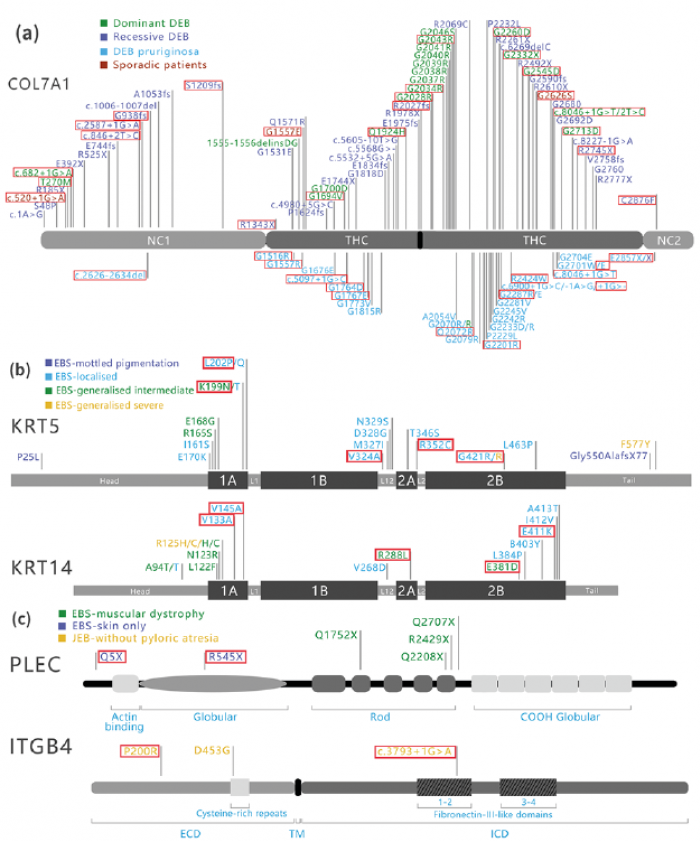
Fig. 1. Mutation spectrum of epidermolysis bullosa (EB) from the Chinese Han patients in COL7A1, KRT5, KRT14, PLEC, and ITGB4 genes. The red highlights are the mutations in the current study. (a) Mutations spectrum of COL7A1 in the Chinese Han EB patients. (b) Mutations spectrum of KRT5 and KRT14 in the Chinese Han EB patients. (c) Mutations spectrum of PLEC and ITGB4 in the Chinese Han EB patients. DEB: dystrophic EB; NC-1: amino-terminal non-collagenous domain; THC: triple helical collagenous domains; NC-2: carboxyl-terminal non-collagenous domain; EBS: EB simplex; L1: linker 1; L1-2: linker 1-2; L2: linker 2; JEB: junctional EB; ECD: extracellular domain; TM: transmembrane domain; ICD: intracellular domain.
Epidermolysis bullosa simplex
Mutations in several genes (KRT5, KRT14, PLEC, KLHL24, EXPH5, CD151 and DST) have been reported in EBS. Most patients with EBS have an AD inheritance pattern, while a few have an AR inheritance pattern (3). In the current study, a total of 12 mutations located in 3 genes (KRT5, KRT14 and PLEC) were identified in 9 pedigrees and 3 sporadic cases of EBS (patients 1–12, Table I). Five mutations in KRT5 were verified in 6 pedigrees, which have been reported previously (5, 6). Five mutations in KRT14 were identified in 5 patients, comprising 2 novel mutations and 3 mutations that were reported previously (5, 6). Case 12, with an AR inheritance pattern, was caused by 2 novel mutations in PLEC.
Based on the new classification of EB (3), 12 patients with EBS were divided into 3 subtypes; 7 EBS-Localized, 4 EBS-Intermediate, and one EBS-Severe (Table I). No correlation between the subtypes of EBS and specific causal genes was found.
Junctional epidermolysis bullosa
JEB is generally inherited in an AR manner and is caused by homozygous or compound mutations in LAMA3, LAMB3, LAMC2, COL17A1, ITGB4, ITGA3 and ITGA6 (3). In this study, a 7-year-old male patient (patient 13) with recessive JEB was found to carry 2 harmful mutations in ITGB4 (Table I). This patient had blistering, scarring, and nail dystrophy. Blistering presented on the buttocks at birth, and the blisters and scars increased to cover the whole body soon after. His urinary system was also affected, which was diagnosed as urethral stricture and cystitis.
Dystrophic epidermolysis bullosa
DEB is caused by mutations in COL7A1, and is categorized into dominant (DDEB) and recessive (RDEB) forms (3). In 44 patients with DEB (patients 14–57), 36 mutations in COL7A1 were found. Twenty-eight DDEB pedigrees carried 5 novel mutations and 8 mutations reported previously (12, 13); 7 RDEB pedigrees carried 3 novel mutations and 11 mutations reported previously (12, 13). The genetic patterns in the 9 remaining sporadic cases were unclear, and 4 novel mutations were found in these patients (Table I).
Patients with DEB presented typical skin fragility, blisters, scars, and nail changes. All 44 patients with DEB comprised 10 DDEB-Localized, 7 DDEB-Intermediate, 1 DDEB-Severe, 10 DDEB-Pruriginosa, 2 RDEB-Intermediate, 4 RDEB-Severe, one RDEB-Pruriginosa, and 9 sporadic cases (Table I) (3). In general, the clinical symptoms of DDEB were milder compared with RDEB. RDEB patient number 40 had a sister who died 60 days after birth, and RDEB patient number 41 had a half-brother who died from blistering and infection at age 2 years. DEB pruriginosa presented the typical manifestations of multiple white papules, nodules, depigmented pigmented spots, and a few scars (14). Among them, 6 patients (patients 42 to 47) presented lesions on their lower limbs, 6 patients (patients 48 to 53) had skin lesions on the lower extremities, and 4 patients (patients 54 to 57) had skin lesions all over their bodies.
WES analysis was used for a relatively large cohort of 57 EB probands and 364 in-house controls in the Chinese population. In this study, 52 mutations in 12 EBS, 1 JEB, and 44 DEB were identified in 5 causal genes, with a mutation detection percentage of 100%. This study emphasized the advantage of WES for accurate diagnosis of patients with EB, and could serve as the basis for future genetic studies on a large-scale to identify and understand EB.
EB is a complex monogenic disorder, with an ever-expanding list of 16 genes known to harbour pathogenic mutations (3). According to the data from different laboratories worldwide, SS has been used to determine that 75–90% of patients with EB carry the responsible mutations (Table II) (5–8). A targeted NGS multi-gene panel and WES have been used to detect mutations underlying EB, with a discovery percentage ranging from 84% to 100% (Table II) (15–20). In the current study, all the mutations of 57 EB pedigrees and sporadic patients in COL7A1, KRT5, KRT14 PLEC, and ITGB4 genes were identified, indicating the value of WES in the genetic diagnosis of Mendelian disease.
Table II. Gene sequencing results of epidermolysis bullosa (EB) in different laboratories
COL7A1 encodes type VII collagen, which is a major component of the anchoring fibrils that anchor the basal lamina to the dermal collagen fibrils. In this study, 36 mutations in COL7A1 were identified in 44 cases (patients 14–57). Based on the literature and the results of our current study in the Chinese Han population, we summarized 102 mutations with 3 subtypes (Fig. 1a).
In DDEB, blisters occur in areas of trauma, typically resulting in scarring, milia formation, and loss of nails. Similar to a previous study in Europeans (19), most patients with DDEB in the current study developed relatively mild clinical symptoms associated with glycine substitutions (GS), which generally occurs in the first position of Gly-X-Y repeats in the triple-helical collagenous (THC) region (Fig. 1a). Dominant GS mutations occur within the THC domain on 1 COL7A1 allele, leading to disruptions of the triple helix structure and the anchor fibres (21). We found 8 GS mutations in exon 73, which was regarded as a “hot exon” of DDEB in previous reports (22). For those missense variants or splice-site alternations, one normal allele can synthesize enough type VII collagen to satisfy the need for anchor fibres, thus DDEB patients with heterozygous mutations in COL7A1 do not blister seriously and have a relatively good quality of life (12). RDEB-severe presents with widespread blisters and scarring, severe mucosal involvement (oral cavity, oesophagus, and anal canal), growth retardation, and pseudosyndactyly of the feet and hands, while RDEB-Localized and RDEB-Generalized have a better prognosis with fewer blisters and mild extracutaneous involvement. Compound heterozygosity is common in RDEB, which is often caused by a combination of mutations, such as premature termination codon (PTC), missense, and splice-site mutations on both alleles (12). PTC mutations decrease the amount of mutated transcripts and lead to truncated nonsense-mediated polypeptides (Fig. 1a) (12, 20). The severe subtypes of RDEB frequently have PTC mutations on both alleles, which causes the complete loss of type VII collagen expression (23). In the current study, patients 37, 40, and 41 carried PTC mutations on both alleles, with severe blistering, abnormal nails, and growth retardation.
DDEB-Pruriginosa and RDEB-Pruriginosa are rare subtypes of DEB caused by COL7A1 (14). In DEB-Pruriginosa, skin fragility, blistering, and scar formation are associated with intense and generalized pruritus, lichenified or nodular prurigo-like lesions, violaceous linear scarring, milia, nail dystrophy, and variable presence of albopapuloid lesions. The current study found 10 DDEB-Pruriginosa pedigrees, one RDEB-Pruriginosa pedigree, and 5 sporadic cases with pruriginosa, which is higher than previous reports (24). In a literature review, mutations in 74 DEB-Pruriginosa patients had been recorded, and 52.7% of cases were caused by GS variants in the THC domain (24), while 9 patients (56%) carried GS variants in our study. Furthermore, the current study found a “hotspot” mutation p.G2287R in 3 patients (patients 51, 52, and 57), which was reported previously (25). All splice site mutations were found in the THC region, and most were located at the 3’end region of exon 87 or near the donor site of intron 87 (Fig. 1a). Four mutations located in this region have been found; c.6899+2A>G, c.6900+1G>C, c.6900+1delG, and c.6900+3G>C in COL7A1 (26).
KRT5 and KRT14 encode keratin 5 and keratin 14, which play an essential role in structural support in basal epidermal cells. Most patients with EBS presenting in early childhood develop blistering on areas of trauma with rare scarring. Patients with EB carrying mutations in KRT5 or KRT14 mostly have AD inheritance forms, but AR has also been reported (3). Mutations in KRT5 and KRT14 destroy the integrity of the keratin intermediate filament network of the basal cells in the epidermis and the stability of the desmosome (6). The current study found 9 patients who carried 8 heterozygous missense mutations in KRT5 and KRT14 with an AD genetic inheritance model. Among them, 7 mutations in these 2 genes were clustered at the 1A and 2B domains. The current study summarized the landscape of 36 mutations in KRT5 and KRT14 in the Chinese population (Fig. 1b). Similar to the worldwide disease-specific database (http://www.interfil.org) and a study in the Netherlands (5–6), 61% (22/36) of mutations in Han Chinese patients occurred in the 1A and 2B domains, and mutations associated with the severe EBS phenotypes commonly clustered at the helix initiation and termination peptide regions in KRT5 and KRT14 (Fig. 1b). The p.R125C/H mutation in KRT14 was previously reported as a “hotspot” for EBS-Severe (8), and the p.P25L mutation in KRT5 was specific for EBS-Mottled pigmentation (27). The 2 mutations above were identified in the Chinese population, suggesting that p.R125C/H and p.P25L were widely prevalent in EBS across different ethnicities in the world.
PLEC, accounting for 8% of EBS in previous reports, encodes plectin, which is ubiquitously present in skin, mucous membranes, gut, muscle, and heart tissue (28). Patient 12 carried 2 novel mutations (p.R559X and p.Q5X) in PLEC with an AR inheritance pattern (Fig. 1c). The blistering of this patient became generalized after birth, while mucous membranes, heart, and muscle were spared. The p.Q5X mutation located in exon 1 encodes plectin isoform 1a (P1a). P1a is widely expressed in the epidermal basal cell layer and cultured keratinocytes; thus, mutations in P1a cause EBS without extracutaneous involvement (29).
Loss-of-function mutations in the ITGB4 gene encoding β4-integrin subunits were identified in JEB-pyloric atresia, which presents with congenital gastrointestinal abnormalities and cutaneous blistering (30). JEB is generally inherited in an AR manner, and approximately 70 patients with JEB with causal ITGB4 mutations have been revealed (31). Patient 13 was diagnosed to have JEB without pyloric atresia, while his urinary system was largely affected. He carried a novel missense mutation p.P200R and a recurrent splice site mutation c.3793+1G>A in ITGB4 (Fig. 1c). The reason for patient 13 having no findings in his urinary system remains to be elucidated.
The current study has some limitations. First, no novel genotype and phenotype relationship was established. Secondly, the limited number of samples included in this study (57 pedigrees and sporadic cases) were not enough to indicate the proportion of each subtype.
In conclusion, comprehensive WES analysis was performed in a cohort of 44 pedigrees and 13 sporadic cases with EB in the Han Chinese population and identified 52 mutations with a percentage of 100%, indicating the power of NGS in diagnosing and classifying EB. The results add novel mutations to the database of the Chinese population with EB, which provide support for prenatal testing and genetic counselling.
The authors sincerely thank the participating patients and the volunteers for their generous and enthusiastic support of this study.
This work was supported by grants from the academic promotion programs of Shandong First Medical University (2019LJ002, 2019RC007, 2020RC001); the Youth Technology Innovation Support Project of Shandong Colleges and Universities (2019KJL003); the Innovation Project of Shandong Academy of Medical Sciences; and the Shandong Province Taishan Scholar Project (tspd20150214, tsqn201812124)
The authors have no conflicts of interest to declare


 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize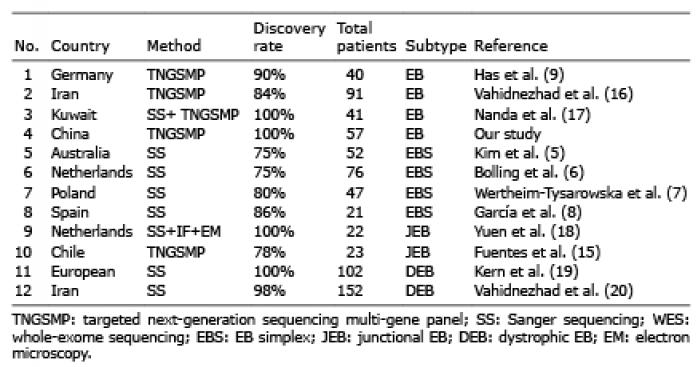 Click to show fullsize
Click to show fullsize